The projects listed below do not currently come with funding. Funding may be sourced at a later date by the supervisor or obtained by the applicant through a funding competition such as Gates, Cambridge Trust or University funding, or other sources of funding.
Exploring Electronic Structure Methods - Dr Alex Thom
In the first instance, please contact Dr Thom's secretary, theory-sec@ ch.cam.ac.uk
Group web site: www.ch.cam.ac.uk/group/thom/
My research focuses on understanding the behaviour of and designing new methods to calculate the electronic structure of molecules. In particular understanding where and why existing methods fail tells us a lot about how to design new, better methods and algorithms. Below I list a number of possible research directions. These potential projects should be suitable for students with a good set of skills in theory and mathematics. A familiarity with computer programming (especially python) would be especially helpful. I am also very happy to discuss potential projects not based on those listed here. These projects are relatively flexible and so can be adapted for relatively short projects from 8 weeks, or extended to form the foundation of a PhD.
Describing Singlet Fission
Owing to its use in light-harvesting materials, there has been much recent interest in the ab initio study of singlet fission. Put simply, light excites an organic molecule into a low-lying excited singlet state, which relaxes into two coupled triplet states (coupled such that the result is still a singlet), which allow charge separation. If harnessed correctly, this is effectively a photovoltaic cell. Two problems generally arise when studying such molecules: i) they must be relatively large for the excited states to be sufficiently low in energy to be accessible with visible light. This makes computational simulation generally expensive; ii) for the excited state to relax to two triplets, it is generally thought that it must be an open-shell singlet. Such states cannot be correctly described by a single Slater determinant, so yet more costly multi-reference methods must be used. In my group, we have been developing methods based on coupling non-orthogonal Slater determinants, and this approach is ideal to describing these open-shell singlet electronic states. This project will investigate the states of polyacene rings and their potential for describing singlet fission processes.
Quantum Chemistry on Quantum Computers
Quantum Computers can in principle solve extremely hard problems in significantly shorter times than conventional algorithms. Within the world of quantum chemistry, both Hartree–Fock and Unitary Coupled Cluster Theory have been used to do calculations on quantum computers, but these are limited in their application by the number of qubits available, which commonly corresponds to the number of orbitals which can be described, and the complexity of the quantum circuits. This project will look at the possibility of using Stochastic Coupled Cluster Theory , and similar algorithms in conjuction with quantum hardware to increase the size of calculations possible.
Holomorphic Density Functional Theory
My group has recently extended the concept of holomorphic Hartree–Fock theoryto a holomorphic Kohn-Sham formalism of Density Functional Theory. This approach allows tracing of so- lutions from Hartree–Fock to KS-DFT with many different functionals. This project will explore the many possibilities that this new theory brings, including how the DFT solutions behave with molecular geometry changes, and investigate whether they are a better approximation to the Full Configuration Interaction Solutions for theses systems.
The Photochemistry of Ethene
The ethene molecule has long been used as a fundamental model for the understanding of photoisomerization processes, and well-studied computationally through the potential energy surfaces of its ground and excited states, usually obtained through CASSCF or MRCI , though recent attempts have been made to study it with density functional theory. The Non-Orthogonal Configuration Interaction Method (NOCI) provides excited states directly from interacting different Hartree–Fock solutions, but does not in itself include dynamical correlation. One possible way to include dynamical correlation is to do so perturbatively. We have derived a NOCI-PT213 to include this dynamical correlation. This project will use NOCI-PT2 to derive a potential energy surface of the ground and excited states of ethene and investigate their properties and look at dynamics on them.
Photochemistry in proteins
Full quantum chemical methods can provide extremely high accuracy energies for ground states along with good estimates of excitation energies of isolated small molecules. However, much photochemistry of interest involves such molecules interacting with a much larger environment, for example a chromophore in a protein scaffold. Unfortunately full quantum chemical methods scale prohibitatively quickly with system size, so cannot be simply applied to such systems, and the general approach is to describe the environment with a lower level of theory to the chromophore, retaining as much of the character of the environment and an accurate description of the molecule of interest. My group has been developing new methods to study excited states via Non-Orthogonal Configuration Interaction14, and this project will investigate whether they can be used effectively in a lower-level protein environment.
Understanding bond-breaking diabatically
A conventional view of a typical mechanism (e.g. an SN2 reaction) involves the simultaneous formation of one bond and breaking of another. In general there is a transition state during this process where there are two partial bonds. An alternative, unconventional view would be that this transition state is the superposition of two different Lewis-bonding configurations, those of the product and those of the reactant. Such states would be the ‘diabatic states’ where the electron configuration is not changed during the process. We are now able to find and track these states (if they indeed exist) during (e.g.) a Hartree–Fock calculation. The transition state may then be formed by a linear combination, using the Non-Orthogonal Configuration Interaction Method (NOCI) . This project will look at some simple reactions and attempt to understand them via this ‘diabatic’ approach.
Understanding Strong Correlation
The Hubbard model is a relatively simple model of strong correlation, with lattice sites (corresponding to a single available orbital on an atom) which can be occupied by electrons, and which shows a rich range of behaviour in its electronic structure. The balance of onsite electron-electron repulsion, U, and kinetic energy, t, determines the nature of the system which moves from metallic for small U/t to insulating for large U/t. Recently, there has been an experimental realization of the Hubbard model with confined cold atoms, and a number of studies showing a breakdown in single-reference correlation techniques at describing such systems. It is likely that symmetry-broken Hartree–Fock solutions will provide a better basis for corrlation treatements. In this project we will investigate the various Hartree–Fock states of the Hubbard model for different U/t, applying correlation treatments to them to understand the origins of their breakdown.
Strong Correlation with Stochastic Coupled Cluster Theory
Conventional Coupled Cluster theory and its stochastic implementation produce highly accurate energies for systems which are dominated by a single electronic configuration, but struggle or fail catastrophically in situations like bond-breadking where there are multiple competing configurations. We have recently extended the stochastic coupled cluster approach to include multiple reference determinants within an active space17 and though this provides a promising starting point for a more general stochastic multireference approach, it is far from black-box for the user. More conventional multireference coupled cluster approaches generally involve a separate Active Space Configuration Interaction calculation to optimize an appropriate active space. This project will explore how an active space can be chosen automatically, and investigate whether a stochastic FCIQMC-like approach can be used within the active space.
High Accuracy Quantum Chemistry for Solids
Conventional wavefunction-based quantum chemistry techniques are becoming increasingly popular when applied to solid state systems owing to their systematic improvability which enables confidence in the results of calculations in areas where techniques like density functional approximations are known not to perform well. These techniques suffer from a poor scaling with system size however rendering only relatively small systems calculable at levels of theory with high accuracy. Coupled Cluster theory, known as the gold standard of Quantum Chemistry, has seldom been applied to solid state systems owing to the infeasability of such calculations. Recently we have developed a stochastic approach to Coupled Cluster theory , which has proven to be able to tackle problems well out of reach of conventional coupled cluster approaches, and has so the potential to tackle problems in solids which are currently infeasible, including in the solid state. This project will investigate whether these calculations on the solid state can accurately represent strong correlation problems such as the Mott– Hubbard insulator transition.
Quantum Astrochemistry
How can we determine the chemical constituents of the stars and planets? Spectroscopy is the obvious answer, but surprisingly experimental data is not usually sufficient in all but the most common molecules. Such projects as ExoMol (www.exomol.com) attempt to draw together this experimental data, but still have limited success. Computational electronic structure techniques should be able to come to the rescue, but the generally available present-day methods do not have the power to predict to the subwavenumber accuracy required. With these highly accurate spectra, the prospect of identifying previously unknown species in exoplanets, space, or even in solar atmospheres becomes feasible. In recent years, members of the department have been developing Quantum Monte Carlo (QMC) techniques which can produce the required accuracy, through highly parallelizable codes. To predict vibrational spectra of a molecule, however, the potential energy surface of such molecules (as well as the dipole moments) is required in order to perform the quantum mechanical calculations on the nuclei in order to generate spectra. This project will combine approaches based on Machine Learning and Gaussian Approximation Potentials19 to the prediction of accurate spectra.
Effect of donor atoms and the crystal field on the single-molecule magnet behaviour of a cobalt and iron complexes: From HartreeFock to multi-reference method
Single-molecule magnets (SMMs) present the smallest possible unit for spin-based electronic devices revolutionising current and future technologies. Although, recent advances in SMMs have shown lanthanide-based complexes to be more promising, the cost and volatility of the rare-earth markets mean that a cheaper and more stable source of materials for SMM fabrication is needed. Independently, Sm and Nd are not popular for SMM while Co and Fe form part of the most interesting transition metal SMM reported. The magnetic properties of powerful classical intermetallics likes SmCo5 and Nd2Fe14B open a new research window for hybrid lanthanide-transition metal SMM design. However, understanding the intrinsic behaviour of each unit separately will form a prerequisite to useful investigation of their hybrids. Smaller systems made up of Co (Figure 2) and Fe could allow us to produce Kramers and non-Kramers ions with insight on the relationship between crystal field effect and minimisation of quantum tunnelling of magnetisation (QTM) in the ground state where breaking of Ms degeneracy and in turn QTM is formally forbidden without field (Figure 1). 20 The crux of the present study will therefore be to investigate factors necessary for the observation of single-molecule magnet behaviour in cobalt and iron complexes using techniques like HF, DFT and ab initio21 multi-reference methods. Figure 1: Double-well energy diagram for negative (left) and positive (right) axial zero-field splitting parameter, D.22 Figure 2: A cobalt(II) complex of 1,2- bis(methanesulfonamido)benzene ligand with tetrathiafulvalenea(TTF) ligand as counterion.
Complex colours
The excited electronic states of molecules play an important role in many parts of chemistry, and as they are necessarily short-lived, they are difficult to study experimentally. Additionally theoretical methods for studying these states are also very limited, and often lack the accuracy to be predictive (e.g. the colours of transition metal complexes are not in any way predictable). Recent research in my group has been investigating using Hartree–Fock Theory to investigate excited electronic states , but such methods are equally applicable to Density Functional Theory, where these excited electronic states have previously been ignored in the theory, as they are difficult to locate. This project will involve investigating the nature of metal-ligand charge transfer transitions and how accurately they can be predicted, comparing multiple DFT solutions with newly developed multireference approached based on multiple Hartree–Fock states (NOCI) . Exploiting reconfigurable hardware Modern high-performace computing systems are usually set up with large numbers of nodes, each with many CPU cores, each node being linked by very fast networks. Recently Graphics Processing Units (GPUs) have begun to be used within such supercomputers, where large amounts of data can be streamed and processed with relatively simple instructions very quickly. Alternative current developments in computer hardware work with hardware such as Field-Programmable Gate Arrays (FPGAs) which can be flashed with a custom circuit design to process streamed data in more complex ways than GPUs. Encoding these designs used to be the speciality of chip designers, but recent innovations have led to compilers which can translate conventionial style programs into circuit designs for such FPGAs, allowing algorithms to be easily converted directly to hardware, with speedups measured in the hundreds when compared to CPUs. This project will investigate encoding Quantum Monte Carlo techniques (which are used for high-accuracy calculations of energies of materials) on FPGAs and the feasability to rework these algorithms to take advantage of new computer architectures, and apply them to some challenging systems, such as solid-state hydrogen phase transitions.
www.ch.cam.ac.uk/group/michaelides/
email: am452@cam.ac.uk
Our research aims at understanding important phenomena in surface-, materials-, and nano-science. Using concepts from quantum mechanics to statistical mechanics, we apply and develop methods and computer simulations to study, for instance, surfaces, interfaces, and processes of environmental relevance. Our work is of both fundamental and applied interest. Water is a major focus of our work. PhD projects are available in the following areas of computational materials science: i) physiochemical properties of solid-liquid interfaces; ii) phase transitions and nucleation; heterogenous catalysis; iv) development and application of high-level electronic structure methods; v) molecular crystals; vi) molecular level studies of olfaction; vii) quantum nuclear effects.
Applying and developing computer simulation methods to better understand environmental ice formation
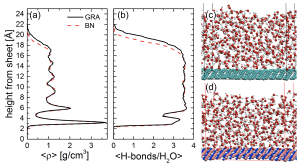
Ice formation is one of the most common phase transitions on Earth. It is relevant to an enormous variety of phenomena such as weathering, cloud formation, airline safety, and energy. However, despite having been studied since antiquity, our molecular level understanding of ice formation is very poor. In particular, almost all ice formation in nature is aided by impurities or the surfaces of foreign materials, yet how surfaces act to facilitate ice formation (heterogeneous ice nucleation) is unclear. Given the ubiquity of ice nucleation, this is arguably one of the biggest unsolved problems in the physical sciences.
This PhD project will focus on applying and developing computer simulation approaches to better understand ice formation on solid substrates. The overall aim is to establish what makes a material good or bad at nucleating ice. The results from this project will not only shed light on an important everyday process but may also help to improve climate models and develop improved cloud seeding materials, or inhibitor coatings for industrial purposes (or maybe even better tasting ice cream!).
Machine learning for the environment – towards improved understanding of confined water and desalination

There is plenty of water on earth but unfortunately most of it is salty and undrinkable. Membranes for purifying and desalinating water exist but there is enormous scope to improve their performance if a clearer understanding of how water interacts with and flows across the surfaces of membrane materials. This project will involve the development and application of state of the art computer simulation approaches to obtain fundamental understanding of water when confined within the pores of membrane mater. The techniques developed and applied will involve a combination of ab initio electronic structure methods and machine learning potentials. Some of the key questions we will seek to answer are: What is the structure of water when confined within the nanometre-scale pores of typical carbon-based membranes? How rapidly can water diffuse across the surfaces of membranes materials? What are the physiochemical factors that optimise water flow versus salt rejection? And more…
Offering projects in the areas of quantum dynamics in the condensed phase. In particular, we are interested in developing methods for simulating reaction dynamics and diffusion on metal surfaces, and computing non-linear infrared spectra for liquid water and ice.
Email address: sca10@cam.ac.uk
Phase behaviour of materials
In our research, we normally use computer simulations and classical statistical mechanics to study the behaviour of various materials, from atomic and molecular to soft condensed matter. In particular, we are interested in nucleation and self-assembly – and combinations of the two – and how thermody- namic and kinetic factors affect and control them. For example, we have investigated in some detail the behaviour of ‘addressable’ self-assembly, in the sense that every molecule in the target structure is unique, which enables extremely finely tuned functionalisation at the nanoscale.
Quantifying bulk phase behaviour requires extensive sampling of phase space, which would be prohibitively expensive using first-principles methods. At the same time, understanding the dynamics of a process from a microscopic perspective is often beyond the reach of experiment. As a result, clever simulation methods are required to be able to study such processes; developing suitable meth- ods to tackle different systems is an interesting research question in which we are interested.
Relevant papers:
• Predicting the phase diagram of titanium dioxide with random search and pattern recognition, Phys. Chem. Chem. Phys., 2020, 22, 12697.
• Direct observation and rational design of nucleation behavior in addressable self-assembly, Proc. Natl Acad. Sci. USA, 2018, 115, E5877.
• Theoretical prediction of thermal polarization, Phys. Rev. Lett., 2018, 120, 226001.
Email: ar732@cam.ac.uk
We are a group of computational scientists fascinated by the study of challenging materials by means of computational methods. We primarily focus on developing machine learning potentials in order to provide insight into complex aqueous systems, for which accurate and efficient representations of potential energy surfaces are urgently needed. Examples of this are the exploration of the behaviour of water at interfaces, or confined in nanotubes and nanochannels, the mechanistic details of ion dissolution and proton transfer in water, as well as the interplay of CO2 and aqueous solutions.
We offer two positions for PhD students this year following the outline below. Funding for one of these projects is available. Please reach out via cs2121@cam.ac.uk to discuss possible research directions and funding options, in particular explaining why you are interested in working in the FAST group alongside a CV.
---
Aqueous Solvation Dynamics on Catalytic Platforms: A Journey Towards A Sustainable Future
In the context of sustainable reactions, the interface between materials and their aqueous surroundings emerges as a pivotal domain.
This PhD project undertakes an exploratory approach to decode the intricate solvation behaviours exhibited by catalytic materials.
These materials such hexagonal boron nitride, MoS2, TiO2, MXenes, or single-atom catalysts on host matrices, strategically positioned for applications such as CO2 reduction and water splitting processes, hold the potential to address climate change challenges.
Leveraging advanced atomistic simulations, this project seeks to unveil the intricate details governing aqueous solvation dynamics in contact with catalytic systems. By dissecting the fundamental interactions at play, the project aims to elucidate the mechanism underlying their catalytic performance. The insights provided through this project will lay a strong foundation to the development of novel sustainable strategies by harnessing the catalytic potential of these materials.
Further examples for interesting reactive processes can be found in “The first-principles phase diagram of monolayer nanoconfined water” Nature, 2022, 609, 512-516.

Figure 1: Nanocatalyst solvated by water
---
Reactions at Solid-Liquid Interfaces through Machine Learning: Efficient Potential Energy Surfaces
Within the evolving landscape of reactions at interfaces, accurate descriptions of surface interactions are of central importance. This PhD project focuses on methodological development, employing machine learning techniques to construct efficient representations of potential energy surfaces. Main challenges in this respect are the accurate inclusion of reactive processes at complex real-world solid-liquid interfaces, as well as the treatment of long-range interactions. Building on state-of-the-art art machine learning potentials based on committee neural networks, the project will explore the inclusion of message passing and physics inspired descriptors to improve the representation of interactions. Additional emphasis will be on the development of workflows for the assembly of training data using active learning strategies to rapidly tackle new systems of interest.
By using machine learning in atomistic simulations, the project will streamline the representation of potential energy landscapes. This advancement aims to unravel the intricate features governing complex solid-liquid interfaces, facilitating a deeper understanding of catalytic reactivity.
The resulting insights will hold the potential to optimise industrial processes, expedite catalyst discovery, and enrich our understanding of processes at interfaces.
Further details on the methodological framework can be found in “Machine learning potentials for complex aqueous systems made simple”, Proc. Nat. Acad. Sci., 2021, 118 (38), e2110077118
Figure 2: Example of a Machine Learning Potential based on a committee of neural networks.